

Why the Ends of the Genome are Important
Why the Ends of the Genome are Important
Discovering what we can learn from simulating shotgun sequencing

Introduction
Two weeks ago, after I posted another installment of my genomic series, I got into an argument with people on Twitter. Before I will post a detailed response, I first want to explain in detail, why in my opinion, it is important to understand why looking at the ends of the genome, in my opinion, is so important.
As outlined before, the original patient sample contained many different sources of RNA (human, other species, viral). Hence, if the authors of Wu et al. 2020, assumed that there was a new virus (or better multiple virions) in the sample, then the full genome should be in the sample.
Overview of my Seven Part Genomics Series:







Fragmentation of RNA
A step in the RNA extraction process, as outlined by Wu et al. 2020, explains the usage of the “SMARTer Stranded Total RNA-Seq kit v.2” by Takara, Inc.
One step of this protocol is to break the total RNA into small pieces for the sequencing step, which can be done via heat or enzymes.

I have contacted the Takara Technical Support, in order to ask “if the position of the cleavage of the RNA in the fragmentation step is random or predetermined?” - here’s their response:

So according to this, the process is resulting in random RNA pieces. Based on this, we can now simulate this process:
Use or generate a reference genome
Fragment into random pieces/reads of ~100bp length
Filter reads of 50-150bp length
Align (or de-novo assemble) the resulting reads back to the reference genome.
Simulated Results
I have written a script to simulate the above process using the SARS-CoV-2 (MN908947.3) reference genome.
My script was configured to start with 100 intact genomes, and a read error rate of 1%. Here are the alignment results of these randomly generated reads (with bwa mem):
Entire Genome

Note, how the reads stack up towards the ends.
Head
Zooming into the head region of the Genome, we can see, that there are 16 reads which perfectly align against the head. There are no random inserts or deletions, and no massive read errors either.

Tail

Same picture here, we can see (at least) 14 reads, that align against the tail perfectly.
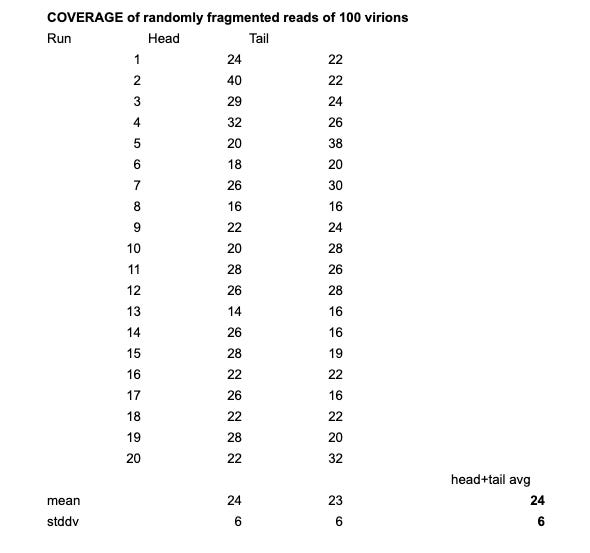
I’ve repeated this step 20x and with 100 virions and 1% read error rate the average depth was 24 reads with a standard deviation of 6.
De-Novo Assembly
Using Megahit to de-novo assemble these reads, consistently results in the (almost) entire genome, if the depth (how many reference genomes are used) is sufficient. In this case, with n=100, we get an almost perfect assembly:
1 contigs, total 29890 bp, min 29890 bp, max 29890 bp, avg 29890 bp, N50 29890 bp
In this case 29890/29903 = 99.99% of the genome was assembled, and only one contig was found, that’s mainly because no other RNA was present in the sample.
The 13 missing bases were missing at the polyA tail, while the head was perfectly reconstructed. Repeating the process with n=1,000 resulted in 6 missing bases at the end, but even with n=10,000 Megahit was not able to assemble the end entirely.
I’ll dive into this into more detail with the next installment of the series.
Summary
So in summary, we could see that using 100 reference genomes, and an error rate of 1%, we end up with about ~15 perfectly aligned reads against the head and the tail (and a general coverage of 24). This means if an entire genome was present in the sample, we should be able to find reads that perfectly align against that novel genome.
Megahit did assemble the head and the ‘body’ of the genome entirely, but struggled with the (polyA)-tail. This could be due to the average read length of 100bp, but the tail just has 34x A’s, so it remains unclear why Megahit is not able to entirely assemble the genome.
I have had deep concerns about PCR since the start. Unfortunately, when I have presented the available evidence, in the form of the Instand report, which showed that positives for SARS-CoV-2 were being triggered from common cold samples, few would listen. Now we are in 2023 and not much has changed: they still deny the results from this report and defend PCR. Same when I show that much of the UK covid PCR testing was testing for one gene - I get barely a shrug. The whole edifice rests on sand.
This work seems very important to me... I hope to have time to study it soon. Bravo for going in this direction! Genomic technology and its limitations must be unpacked. It has captured too much territory.